In vitro Transcription: mRNA Therapeutics
How to design mRNA therapeutics for scalable success
The recent Nobel Prize awarded to Katalin Karikó and Drew Weissman underlined the impact their breakthroughs in mRNA technology had in combating COVID-19. This new class of therapeutics is poised to have an increasingly important role in the future of healthcare.
This blog post outlines some of the considerations for the design, production and purity testing of mRNA therapeutic programmes.
Although the Pfizer and Moderna COVID-19 vaccines were the first mRNA technology-based medicines to be approved by regulators, by 2022 there were five new mRNA therapeutics in clinical development stages registered with the FDA. With trials targeting ischemic heart disease, advanced melanoma and cystic fibrosis, mRNA therapeutics are a fast-emerging platform offering hope for a wide range of diseases.

From being a promising yet under explored approach in the 1990s, mRNA therapeutics have evolved significantly because chemical modifications have helped to overcome the challenges of instability and immunotoxicity. The scalability, potential for rapid production, and relatively straightforward manufacturing process give mRNA-based therapeutics an advantage over traditional drugs and DNA technology. However, there are many factors that are crucial for delivering a successful mRNA therapeutic.
The mRNA production process
mRNA is manufactured by a simple in-vitro enzymatic process involving upstream and downstream steps.
Upstream mRNA processing
The upstream production process includes the enzymatic generation of mRNA. It has several steps: plasmid production, plasmid linearisation, in vitro transcription, 5′ capping and adding a poly(A) tail.
Specific RNA polymerases, including T7, SP6 or T3, catalyse target mRNA synthesis from a corresponding DNA template. Generally, the DNA template is produced by linearisation of a purified plasmid or by using PCR to amplify a region of interest.
In addition, in-vitro transcription (IVT) requires nucleoside triphosphates (NTPs), a pH-controlling buffer and a polymerase cofactor. The IVT reaction is complete within a few hours, compared to the lengthy processes involved in conventional vaccine production. This simpler manufacturing process could help to reduce contamination risks.
mRNA transcription, capping and poly(A) tail addition can combine into one, two or three steps depending on the synthesis route design.
mRNA capping can be achieved during transcription using cap analogues or post-transcriptionally via a second enzymatic reaction using a vaccinia capping enzyme. The benefit of using cap analogues is a faster process that doesn’t require a separate reaction step. However, capping efficiency is higher using the two-step capping method. mRNA therapeutics producers should complete a thorough risk analysis to identify the most appropriate capping strategy for their application.
A poly(A) tail can be added during transcription by inserting the tail sequence into the DNA template. Alternatively, it may be added post-transcriptionally via a separate enzymatic reaction using poly(A) polymerase. Using the enzyme allows users to create a longer poly(A) tail and therefore increase the stability of the mRNA.
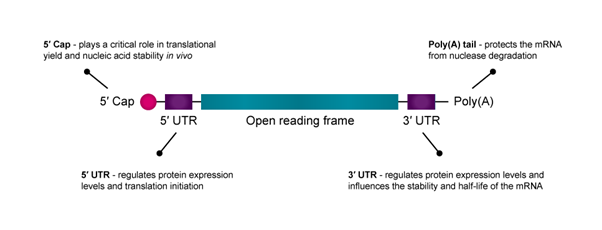
Figure 1: An illustration of the structure of mRNA with key elements highlighted.
Downstream mRNA processing
The downstream production process purifies the final mRNA product. Impurities, including enzymes, residual NTPs, DNA templates and unwanted mRNAs, must be removed to achieve clinical purity standards.
Generally, an endonuclease for DNase digestion is used to remove undesirable DNA, followed by lithium chloride precipitation. Chromatography is widely used in the biotechnology industry for nucleic acid purification as it is selective, versatile, scalable, cost-effective and removes aberrant mRNA, including dsRNA and truncated RNA fragments. Researchers have observed a 10–1000-fold increase in protein production when nucleoside-modified mRNA was purified by reverse-phase high-performance liquid chromatography prior to cell delivery.
It is possible to assess the efficacy of mRNA capping and/or eliminate undesirable, uncapped RNAs using RNA 5′ Polyphosphatase and Terminator™ 5'-Phosphate-Dependent Exonuclease. RNA 5′ Polyphosphatase is an Mg2+-independent phosphohydrolase that sequentially removes the γ and β phosphates from 5′-triphosphorylated RNA (such as primary RNA transcripts). RNA Polyphosphatase has no activity on RNA with a 5′ cap and is useful in preparing substrate RNA molecules for subsequent degradation using the Terminator 5′-Phosphate-Dependent Exonuclease.
Ensuring mRNA purity and integrity

The purity and integrity of mRNA are critical, as impurities may decrease translational efficiency or increase immunogenicity risks. Partnering with preferred suppliers that guarantee the products meet ISO 13485 standards can ensure that enzymes for the IVT reaction are high-quality and reliable.
Potential consequences for using sub-optimal raw materials include:
• Enhanced risk for workflow delays due to lot-to-lot variation in enzyme performance
• Additional purification increases production costs
• Potential for non-conformance and supplier qualification issues during regulatory review by Competent Authorities
• Difficulty scaling production for commercialisation
• Wasted time and resources on repeat work due to variability in raw material performance
mRNA design for delivery and immunogenicity
Cell delivery and immunogenicity are two challenges associated with mRNA therapeutics. To overcome these obstacles, tailored mRNA design and synthesis are critical for therapeutic efficacy.
mRNA consists of five functional regions, including the 5′ cap, the 3′ poly(A) tail, the open reading frame (ORF) flanking and the 3′ untranslated regions (UTRs), which mediate the translation efficacy and decay rate of mRNA.
Cap structure
The 5′ cap of mRNA plays a critical role in translational yield and nucleic acid stability in vivo. Because translation is often rate-limited by the cap-dependent initiation process, optimising the 5′ cap can improve mRNA drug efficacy and stability.2 Research also shows that modifying the first nucleotide after the 5′-cap may increase transcript stability and translational yield.
Examples of 5′ cap modifications include:3
• Phosphodiester analogues
• “Anti-reverse” caps (2’- and 3’ OH modifications)
• First nucleotide identity and methylation status (cap 0, 1, 2)
These modifications can help:
• Increase translational efficiency via binding affinity to eIF4E
• Improve mRNA lifespan by resisting decapping
• Avoid detection by innate immune sensors

UTRs
The untranslated regions (UTRs) at the 5′ and 3′ terminals of mRNA are important to optimise for enhancing protein expression.3 While both the 5′-UTR and 3′-UTR regulate protein expression levels, the 5′-UTR is primarily responsible for translation initiation, and the 3′-UTR influences the stability and half-life of the mRNA.
Longer 3′ UTRs are associated with shorter mRNA half-life, while shorter 3′ UTRs reduce translation efficiency. 3′ UTRs derived from α- and β-globins are commonly used in mRNA therapeutics for delivery into various cell types. However, researchers continue to screen potential UTRs to further optimise protein expression and cell delivery.
Examples of UTR modifications include:
• Full nucleotide replacement
• Context-dependent modified nucleosides
• Phosphodiester analogues
These modifications can help:
• Suppress immune detection, especially when uridine substitution is employed
• Improve mRNA stability and translational yield
Poly(A) tail
An mRNA construct’s poly(A) tail protects it from nuclease degradation. Generally, longer poly(A) tails improve mRNA stability and translation, although the target cell type will largely determine the optimal tail size.
Examples of poly(A) tail modifications include:
• Exonuclease resistant backbones
• Conjugated fluorescent reporters
These modifications can help:
• Increase mRNA stability by resisting exonucleolytic digestion
• Improve translational efficiency
Designing mRNA-based therapeutics for practical application
Beyond the biological challenges associated with mRNA therapeutics, developers must also address storage, clinical application and regulatory hurdles.
For instance, the SARS-CoV-2 mRNA vaccines require ultra-cold storage conditions that may not be feasible in developing countries. The cost to ship and store these temperature-sensitive therapeutics must be a consideration for manufacturers.
Developing thermostable products with high clinical efficacy remains a vast opportunity to advance mRNA therapeutics. Research has shown that there are ways to improve the thermostability of mRNA therapeutics, including lyophilisation, mRNA sequence optimisation, and optimisation of the components and manufacturing process of nanoparticles, including LNP. Short shelf life is also a concern for mRNA therapeutics and can be a limiting factor for clinical application.

With the rapid pace of advances in mRNA therapeutic technology, the regulatory pathway is having to adapt to keep up. Moving a product from the discovery to the commercial phase will require proactive conversations with regulatory stakeholders to ensure that raw material selection, construct design, data capture and manufacturing processes meet expectations for clinical applications.
What lies ahead?
As we navigate the frontier of mRNA therapeutics, the potential for transforming disease management and treatment is vast. The need to consider the end purpose is vital when navigating the choices in manufacturing, design and raw materials in order to clear the practical hurdles before reaching the clinic.
Circular RNA (circRNA) is the next generation of mRNA therapy and presents an opportunity to enhance pharmaceutical stability and biostability compared to linear mRNA therapeutics. Current challenges associated with linear mRNA therapeutics, including exonuclease degradation, are alleviated by the covalently closed structures of circRNA. The next blog post of this series will take a closer look at circRNA and its role in the future of mRNA therapeutics.
Our range of Enzymes and kits for IVT mRNA synthesis and RNA therapeutics
- Prevent RNA degradation with RiboGuard™ RNase Inhibitor.
- Digest the DNA template with RNase-Free DNase I.
- Add poly(A) tails to the 3′ ends of IVT RNAs with Poly(A) polymerase tailing kit.
- Post-transcriptional capping with ScriptCap™Capping systems
- Digests all linear RNAs but lariat or circular RNA with RNase R
- Remove interfering DNA from RNA preps with RNase-Free DNase I
- Removal of genomic small and DNA - Baseline-ZERO™ DNase
- Circularise your DNA template with -CircLigase™ DNA Ligase